Physics in high dimensions is becoming the norm in modern dynamics. It is not only that string theory operates in ten dimensions (plus one for time), but virtually every complex dynamical system is described and analyzed within state spaces of high dimensionality. Population dynamics, for instance, may describe hundreds or thousands of different species, each of whose time-varying populations define a separate axis in a high-dimensional space. Coupled mechanical systems likewise may have hundreds or thousands (or more) of degrees of freedom that are described in high-dimensional phase space.
In high-dimensional landscapes, mountain ridges are much more common than mountain peaks. This has profound consequences for the evolution of life, the dynamics of complex systems, and the power of machine learning.
For these reasons, as physics students today are being increasingly exposed to the challenges and problems of high-dimensional dynamics, it is important to build tools they can use to give them an intuitive feeling for the highly unintuitive behavior of systems in high-D.
Within the rapidly-developing field of machine learning, which often deals with landscapes (loss functions or objective functions) in high dimensions that need to be minimized, high dimensions are usually referred to in the negative as “The Curse of Dimensionality”.
Dimensionality might be viewed as a curse for several reasons. First, it is almost impossible to visualize data in dimensions higher than d = 4 (the fourth dimension can sometimes be visualized using colors or time series). Second, too many degrees of freedom create too many variables to fit or model, leading to the classic problem of overfitting. Put simply, there is an absurdly large amount of room in high dimensions. Third, our intuition about relationships among areas and volumes are highly biased by our low-dimensional 3D experiences, causing us to have serious misconceptions about geometric objects in high-dimensional spaces. Physical processes occurring in 3D can be over-generalized to give preconceived notions that just don’t hold true in higher dimensions.
Take, for example, the random walk. It is usually taught starting from a 1-dimensional random walk (flipping a coin) that is then extended to 2D and then to 3D…most textbooks stopping there. But random walks in high dimensions are the rule rather than the exception in complex systems. One example that is especially important in this context is the problem of molecular evolution. Each site on a genome represents an independent degree of freedom, and molecular evolution can be described as a random walk through that space, but the space of all possible genetic mutations is enormous. Faced with such an astronomically large set of permutations, it is difficult to conceive of how random mutations could possibly create something as complex as, say, ATP synthase which is the basis of all higher bioenergetics. Fortunately, the answer to this puzzle lies in the physics of random walks in high dimensions.
Why Ten Dimensions?
This blog presents the physics of random walks in 10 dimensions. Actually, there is nothing special about 10 dimensions versus 9 or 11 or 20, but it gives a convenient demonstration of high-dimensional physics for several reasons. First, it is high enough above our 3 dimensions that there is no hope to visualize it effectively, even by using projections, so it forces us to contend with the intrinsic “unvisualizability” of high dimensions. Second, ten dimensions is just big enough that it behaves roughly like any higher dimension, at least when it comes to random walks. Third, it is about as big as can be handled with typical memory sizes of computers. For instance, a ten-dimensional hypercubic lattice with 10 discrete sites along each dimension has 10^10 lattice points (10 Billion or 10 Gigs) which is about the limit of what a typical computer can handle with internal memory.
As a starting point for visualization, let’s begin with the well-known 4D hypercube but extend it to a 4D hyperlattice with three values along each dimension instead of two. The resulting 4D lattice can be displayed in 2D as a network with 3^4 = 81 nodes and 216 links or edges. The result is shown in Fig. 1, represented in two dimensions as a network graph with nodes and edges. Each node has four links with neighbors. Despite the apparent 3D look that this graph has about it, if you look closely you will see the frustration that occurs when trying to link to 4 neighbors, causing many long-distance links.
[See YouTube video for movies showing evolving hyperlattices and random walks in 10D.]

We can also look at a 10D hypercube that has 2^10 = 1024 nodes and 5120 edges, shown in Fig. 2. It is a bit difficult to see the hypercubic symmetry when presented in 2D, but each node has exactly 10 links.

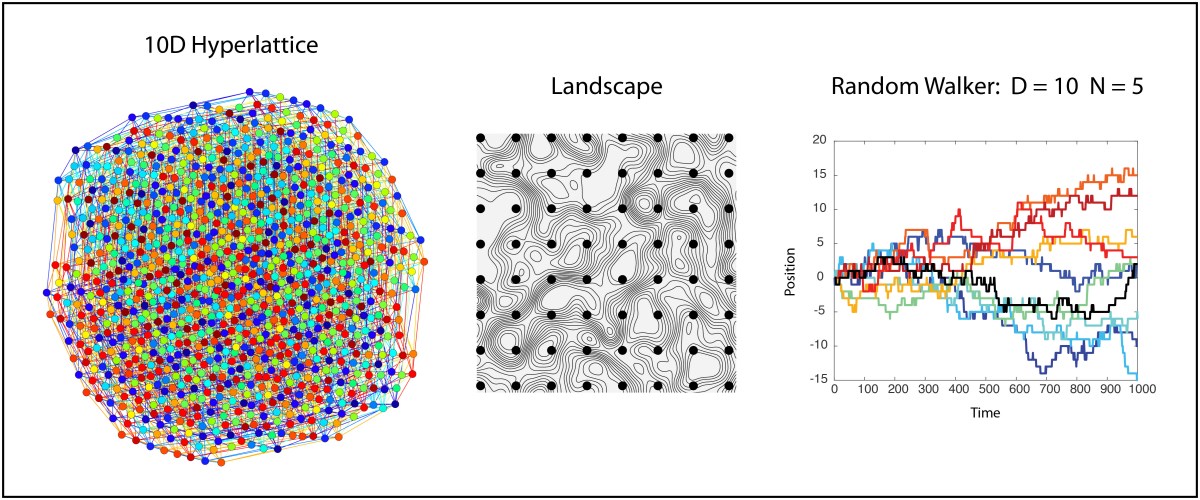
Extending this 10D lattice to 10 positions instead of 2 and trying to visualize it is prohibitive, since the resulting graph in 2D just looks like a mass of overlapping circles. However, our interest extends not just to ten locations per dimension, but to an unlimited number of locations. This is the 10D infinite lattice on which we want to explore the physics of the random walk.
Diffusion in Ten Dimensions
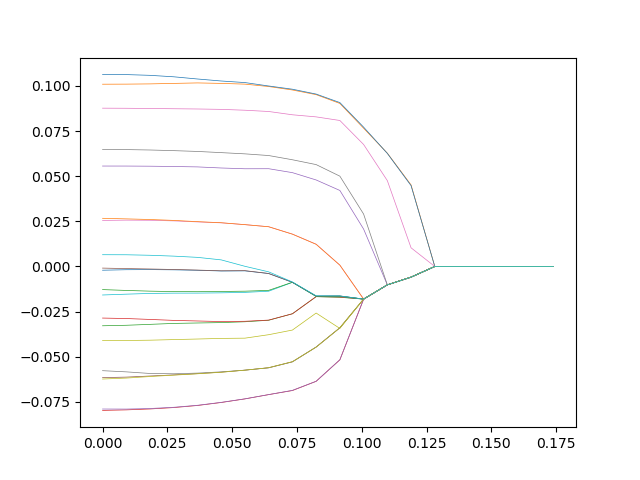
An unconstrained random walk in 10D is just a minimal extension beyond a simple random walk in 1D. Because each dimension is independent, a single random walker takes a random step along any of the 10 dimensions at each iteration so that motion in any one of the 10 dimensions is just a 1D random walk. Therefore, a simple way to visualize this random walk in 10D is simply to plot the walk against each dimension, as in Fig. 3. There is one chance in ten that the walker will take a positive or negative step along any given dimension at each time point.

An alternate visualization of the 10D random walker is shown in Fig. 4 for the same data as Fig. 3. In this case the displacement is color coded, and each column is a different dimension. Time is on the vertical axis (starting at the top and increasing downward). This type of color map can easily be extended to hundreds of dimensions. Each row is a position vector of the single walker in the 10D space

In the 10D hyperlattice in this section, all lattice sites are accessible at each time point, so there is no constraint preventing the walk from visiting a previously-visited node. There is a possible adjustment that can be made to the walk that prevents it from ever crossing its own path. This is known as a self-avoiding-walk (SAW). In two dimensions, there is a major difference in the geometric and dynamical properties of an ordinary walk and an SAW. However, in dimensions larger than 4, it turns out that there are so many possibilities of where to go (high-dimensional spaces have so much free room) that it is highly unlikely that a random walk will ever cross itself. Therefore, in our 10D hyperlattice we do not need to make the distinction between an ordinary walk and a self-avoiding-walk. However, there are other constraints that can be imposed that mimic how complex systems evolve in time, and these constraints can have important consequences, as we see next.
Random Walk in a Maximally Rough Landscape
In the infinite hyperlattice of the previous section, all lattice sites are the same and are all equally accessible. However, in the study of complex systems, it is common to assign a value to each node in a high-dimensional lattice. This value can be assigned by a potential function, producing a high-dimensional potential landscape over the lattice geometry. Or the value might be the survival fitness of a species, producing a high-dimensional fitness landscape that governs how species compete and evolve. Or the value might be a loss function (an objective function) in a minimization problem from multivariate analysis or machine learning. In all of these cases, the scalar value on the nodes defines a landscape over which a state point executes a walk. The question then becomes, what are the properties of a landscape in high dimensions, and how does it affect a random walker?
As an example, let’s consider a landscape that is completely random point-to-point. There are no correlations in this landscape, making it maximally rough. Then we require that a random walker takes a walk along iso-potentials in this landscape, never increasing and never decreasing its potential. Beginning with our spatial intuition living in 3D space, we might be concerned that such a walker would quickly get confined in some area of the lanscape. Think of a 2D topo map with countour lines drawn on it — If we start at a certain elevation on a mountain side, then if we must walk along directions that maintain our elevation, we stay on a given contour and eventually come back to our starting point after circling the mountain peak — we are trapped! But this intuition informed by our 3D lives is misleading. What happens in our 10D hyperlattice?
To make the example easy to analyze, let’s assume that our potential function is restricted to N discrete values. This means that of the 10 neighbors to a given walker site, on average only 10/N are likely to have the same potential value as the given walker site. This constrains the available sites for the walker, and it converts the uniform hyperlattice into a hyperlattice site percolation problem.
Percolation theory is a fascinating topic in statistical physics. There are many deep concepts that come from asking simple questions about how nodes are connected across a network. The most important aspect of percolation theory is the concept of a percolation threshold. Starting with a complete network that is connected end-to-end, start removing nodes at random. For some critical fraction of nodes removed (on average) there will no longer be a single connected cluster that spans the network. This critical fraction is known as the percolation threshold. Above the percolation threshold, a random walker can get from one part of the network to another. Below the percolation threshold, the random walker is confined to a local cluster.
If a hyperlattice has N discrete values for the landscape potential (or height, or contour) and if a random walker can only move to site that has the same value as the walker’s current value (remains on the level set), then only a fraction of the hyperlattice sites are available to the walker, and the question of whether the walker can find a path the spans the hyperlattice becomes simply a question of how the fraction of available sites relates to the percolation threshold.
The percolation threshold for hyperlattices is well known. For reasonably high dimensions, it is given to good accuracy by

where d is the dimension of the hyperlattice. For a 10D hyperlattice the percolation threshold is pc(10) = 0.0568, or about 6%. Therefore, if more than 6% of the sites of the hyperlattice have the same value as the walker’s current site, then the walker is free to roam about the hyperlattice.
If there are N = 5 discrete values for the potential, then 20% of the sites are available, which is above the percolation threshold, and walkers can go as far as they want. This statement holds true no matter what the starting value is. It might be 5, which means the walker is as high on the landscape as they can get. Or it might be 1, which means the walker is as low on the landscape as they can get. Yet even if they are at the top, if the available site fraction is above the percolation threshold, then the walker can stay on the high mountain ridge, spanning the landscape. The same is true if they start at the bottom of a valley. Therefore, mountain ridges are very common, as are deep valleys, yet they allow full mobility about the geography. On the other hand, a so-called mountain peak would be a 5 surrounded by 4’s or lower. The odds for having this happen in 10D are 0.2*(1-0.8^10) = 0.18. Then the total density of mountain peaks, in a 10D hyperlattice with 5 potential values, is only 18%. Therefore, mountain peaks are rare in 10D, while mountain ridges are common. In even higher dimensions, the percolation threshold decreases roughly inversely with the dimensionality, and mountain peaks become extremely rare and play virtually no part in walks about the landscape.
To illustrate this point, Fig. 5 is the same 10D network that is in Fig. 2, but only the nodes sharing the same value are shown for N = 5, which means that only 20% of the nodes are accessible to a walker who stays only on nodes with the same values. There is a “giant cluster” that remains connected, spanning the original network. If the original network is infinite, then the giant cluster is also infinite but contains a finite fraction of the nodes.

The quantitative details of the random walk can change depending on the proximity of the sub-networks (the clusters, the ridges or the level sets) to the percolation threshold. For instance, a random walker in D =10 with N = 5 is shown in Fig. 6. The diffusion is a bit slower than in the unconstrained walk of Figs. 3 and 4. But the ability to wander about the 10D space is retained.

This is then the general important result: In high-dimensional landscapes, mountain ridges are much more common than mountain peaks. This has profound consequences for the evolution of life, the dynamics of complex systems, and the power of machine learning.
Consequences for Evolution and Machine Learning
When the high-dimensional space is the space of possible mutations on a genome, and when the landscape is a fitness landscape that assigns a survival advantage for one mutation relative to others, then the random walk describes the evolution of a species across generations. The prevalence of ridges, or more generally level sets, in high dimensions has a major consequence for the evolutionary process, because a species can walk along a level set acquiring many possible mutations that have only neutral effects on the survivability of the species. At the same time, the genetic make-up is constantly drifting around in this “neutral network”, allowing the species’ genome to access distant parts of the space. Then, at some point, natural selection may tip the species up a nearby (but rare) peak, and a new equilibrium is attained for the species.
One of the early criticisms of fitness landscapes was the (erroneous) criticism that for a species to move from one fitness peak to another, it would have to go down and cross wide valleys of low fitness to get to another peak. But this was a left-over from thinking in 3D. In high-D, neutral networks are ubiquitous, and a mutation can take a step away from one fitness peak onto one of the neutral networks, which can be sampled by a random walk until the state is near some distant peak. It is no longer necessary to think in terms of high peaks and low valleys of fitness — just random walks. The evolution of extremely complex structures, like ATP synthase, can then be understood as a random walk along networks of nearly-neutral fitness — once our 3D biases are eliminated.
The same arguments hold for many situations in machine learning and especially deep learning. When training a deep neural network, there can be thousands of neural weights that need to be trained through the minimization of a loss function, also known as an objective function. The loss function is the equivalent to a potential, and minimizing the loss function over the thousands of dimensions is the same problem as maximizing the fitness of an evolving species.
At first look, one might think that deep learning is doomed to failure. We have all learned, from the earliest days in calculus, that enough adjustable parameter can fit anything, but the fit is meaningless because it predicts nothing. Deep learning seems to be the worst example of this. How can fitting thousands of adjustable parameters be useful when the dimensionality of the optimization space is orders of magnitude larger than the degrees of freedom of the system being modeled?
The answer comes from the geometry of high dimensions. The prevalence of neutral networks in high dimensions gives lots of chances to escape local minima. In fact, local minima are actually rare in high dimensions, and when they do occur, there is a neutral network nearby onto which they can escape (if the effective temperature of the learning process is set sufficiently high). Therefore, despite the insanely large number of adjustable parameters, general solutions, that are meaningful and predictive, can be found by adding random walks around the objective landscape as a partial strategy in combination with gradient descent.
Given the superficial analogy of deep learning to the human mind, the geometry of random walks in ultra-high dimensions may partially explain our own intelligence and consciousness.
Biblography
S. Gravilet, Fitness Landscapes and the Origins of Species. Princeton University Press, 2004.
M. Kimura, The Neutral Theory of Molecular Evolution. Cambridge University Press, 1968.